肝豆状核变性有不发病的患者。
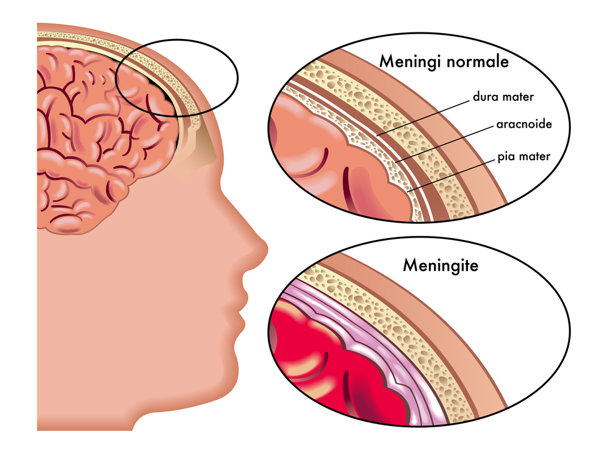
肝豆状核变性是由ATP7B基因突变引起的遗传代谢疾病,其致病机制涉及到铜离子的代谢和排泄过程中的遗传缺陷。对于没有突变的个体而言,正常的铜代谢途径得以维持,因此不会出现铜在体内的异常积累,也就不会表现出疾病的典型症状。
肝豆状核变性可能因不同的遗传机制而存在多种亚型,其中某些亚型可能存在较低的发病率或较轻的症状表现。
针对肝豆状核变性的诊断与管理需特别考虑家族史,并定期监测患者的铜蓝蛋白水平以及神经系统功能。

若患者无家族史且铜蓝蛋白水平正常,则可排除该疾病的可能性。